
This semester, alongside our long-standing systems research, we will explore practical ways to pair our work with LLMs. For example, improving developer documentation, identifying code invariants, visualizing logs, and troubleshooting workflows. Ground rules: you are responsible for your output; do not create extra work for others; use ND-authorized tools such as Gemini and Gemini-CLI to protect data. This week each person will pick a modest task and tool to explore. Next week we will report on methods, results, and observations.

Recent News in the CCL
Reducing Overhead of LLM-integrated Applications on GPU Clusters with Parsl+TaskVine
Large Language Models (LLMs) are becoming a key tool for scientific discovery, but using them on High-Performance Computing (HPC) clusters is challenging due to the limitations of traditional resource allocation methods. For instance, static allocation, which assigns a dedicated set of GPUs for a task, is a rigid system. This can lead to long queues of frustrated users and wasted resources, as the allocated GPUs sit idle while waiting for the next job to start. Meanwhile, opportunistic allocation allows tasks to use available, but not guaranteed, resources. While this improves overall cluster utilization, it's problematic for LLM applications. The initial loading of a multi-billion-parameter LLM is a time-consuming process, and since tasks in an opportunistic environment can be preempted at any moment, this expensive startup often has to be repeated from scratch.

To solve this, we propose a new technique called Pervasive Context Management. The core idea is to decouple the LLM initialization context from the actual inference tasks and keep this context persistent on GPUs until it is no longer needed. This transforms the high startup cost into a one-time, amortizable expense. When a task is preempted, it can be quickly rescheduled to another GPU that already has the necessary context loaded, eliminating the need to re-initialize the model. Our Parsl+TaskVine system can also transfer existing context between nodes to bootstrap new GPUs, reducing data transfer time and avoiding bottlenecks.

To demonstrate the effectiveness of this approach, we transformed a fact-verification application to use Pervasive Context Management and conducted a comprehensive evaluation. Our results show a significant improvement in performance on both static and opportunistic resources. By enabling Pervasive Context Management, the end-to-end execution time of our application was reduced by 72.1%, from 3 hours to just 48 minutes, using the same number of GPUs. The application also scaled efficiently on up to 32.8% of all GPUs (up to 186 GPUs) in the cluster, further reducing the execution time to a mere 13 minutes.

Additionally, Pervasive Context Management helps users avoid the complex problem of tuning the inference batch size. Because the expensive startup cost is now a one-time event, the application's performance becomes much more stable regardless of the batch size chosen. This removes the burden of manual tuning and ensures near-optimal execution. In summary, our findings show that this new approach is a viable solution for running high-throughput LLM inference applications efficiently on heterogeneous and opportunistic HPC clusters.
TaskVine Insights: Storage Management – PFS vs. NLS
There are two primary storage layers when running workflows in HPC environments:
-
Parallel File System (PFS): A shared file system accessible to all users in the same cluster, such as VAST, Lustre, BeeGFS, and CephFS.
-
Node-Local Storage (NLS): Each worker’s local disk, accessed directly without relying on the network, usually the temporary directory of the local file system.

PFS and NLS each have their own advantages and disadvantages.
PFS is convenient because it can be easily accessed by users. It usually has large capacity, often hundreds of terabytes, making it an ideal choice for storing big datasets. It is also stable and reliable, ensuring that data is not lost. However, the main drawback of PFS is that its I/O bandwidth is shared among many users, so it can become saturated when many jobs perform I/O at the same time, turning data access into a major bottleneck for parallel computation.
In contrast, NLS provides isolated storage on distributed workers. Each worker has its own local disk and can read and write data directly. This allows the total I/O bandwidth to aggregate across all workers, and data transfer between nodes happens through peer-to-peer communication. This design effectively reduces the I/O contention that occurs on the PFS and helps workflows scale to larger sizes. On the flip side, it also has limitations. Its capacity is small, typically a few hundred gigabytes per node, and it is less reliable, because node failures are unpredictable, and when a node is preempted or goes offline, all data stored on that node is lost, which poses a risk to users.
The figure below shows the performance difference between PFS and NLS. We test concurrent reads and writes from 1 to 128 threads and measure their average bandwidth. The PFS test runs directly on the parallel file system, while the NLS test runs on 8 workers, each with 16 cores. It is clear that running parallel I/O on NLS provides much higher average bandwidth. Each thread maintains about 1 GB/s of throughput, whereas the average bandwidth on PFS drops sharply as concurrency increases.

The key takeaway from this comparison is that running large-scale or data-intensive workflows on HPC systems requires relying on NLS for its high aggregate I/O bandwidth, ensuring that the entire workflow is not slowed down by a deluge of reads and writes.
Our team has been studying and improving how to better leverage NLS to accelerate large-scale workflow computations in HPC systems through TaskVine, a workflow management system we have been developing over the past few years. TaskVine’s key advantage is its ability to use each worker’s NLS to reduce I/O contention on the PFS, enabling faster data access and quicker workflow completion. It also employs a range of data management techniques and strategies to ensure that NLS is used effectively and efficiently, keeping data safe and handling unpredictable node failures with care.
This blog series shares how we manage data carefully, address the challenges of using NLS when local disk space is limited and nodes are prone to failures, and achieve massive scalability.
Stay tuned for upcoming technical insights, code examples, and updates!
Tue, 07 Oct 2025 18:06:00 +0000eScience 2025: Liberating the Data Aware Scheduler to Achieve Locality in Layered Scientific Workflow Systems
On September 16 graduate student Colin Thomas presented the paper titled: Liberating the Data Aware Scheduler to Achieve Locality in Layered Scientific Workflow Systems at the 21st IEEE International Conference on eScience in Chicago, Illinois.
This work engages mutliple topics including workflow systems, data management, and task scheduling. The title describes two key components; The Data Aware Scheduler, and Layered Scientific Workflow Systems. Data aware schedulers are capable of understanding task data dependencies and making task scheduling decisions based on that information, primarily to benefit from data locality when there is intermediate workflow data which is already cached somewhere in the cluster. It is beneficial to schedule tasks who consume this data to the same site in which the data was created, so that the dependencies to not have to move through the network, or perhaps even out of memory.
The term "layered workflow system" is a way to describe multiple popular workflow systems in the HPC community such as Parsl and Dask. These workflow systems consist of two primary components. The DAG manager and executor. The DAG manager understands the workflow composition and data dependencies. The executor receives tasks from the DAG manager and uses its understanding of the cluster and available resources to place tasks on their execution sites.
The primary argument of the paper is highlighting the obstacles created by using a data aware scheduler in this layered execution scheme. If we take a DAG such as the one described by Figure 1, we can easily identify opportunities for data locality in the groups of sequentially dependent tasks. However the data aware scheduler is not privy to this picture of the DAG. Rather the scheduler, or executor, is only aware of tasks which are ready to run, while the DAG manager withholds future task information until they are ready to run as well. This forces a data aware scheduler to make last-minute scheduling decisions based on individual tasks. In many cases it may occupy a node in which a later task would have been better suited due to data locality opportunities.
The paper shows an implementation of a modified Parsl DAG manager and TaskVine data aware scheduler which passes through intermediate-dependent sequential tasks to the data aware scheduler before all of them are ready to run. This allows TaskVine to identify the group dependencies and the ideal execution pattern, and schedule these task groups in batches rather than on an individual basis.
The result of this increases the data locality achieved on the 2 workflows in the evaluation. In addition it reduces the total number of scheduling operations by a factor of the average task group size.
The link to the full paper can be found below:
https://ccl.cse.nd.edu/research/papers/liberating-escience-2025.pdf
Wed, 01 Oct 2025 14:55:00 +0000
Floability at eScience 2025: Making Notebooks Portable with Backpacks Across HPC Clusters
Grad student Saiful Islam presented our paper “Backpacks for Notebooks: Enabling Containerized Notebook Workflows in Distributed Environments” at the 2025 IEEE eScience Conference in Chicago, Illinois.
Notebooks have become the de facto interface for scientific computing, but moving from a local notebook to large-scale HPC clusters is far from seamless—especially when the notebook contains a distributed workflow that must be submitted across multiple nodes. A notebook file alone doesn’t capture the full execution context—it misses environment specifications, data locations, and resource requirements. As a result, the same notebook often runs on one system but fails on another.

This paper introduces the concept of a backpack—a lightweight companion that travels with the notebook and captures everything needed for execution. A backpack makes explicit the software environment, data sources, and resource requirements that are often left implicit in code. With Floability, our implementation of backpack specifications, backpacks transform ordinary notebooks into portable, reproducible workflows that can execute across heterogeneous HPC clusters with zero to minimal code modification.

We evaluated Floability on three representative scientific workflows—distributed image convolution, climate trend analysis, and high-energy physics data analysis—running them across five heterogeneous HPC systems (Notre Dame CRC, Purdue Anvil, UT Stampede3, OSPool, and AWS). In each case, backpacks successfully captured the required software and data dependencies, provisioned worker environments, and reproduced execution without code changes. While runtime varied due to site-specific infrastructure like schedulers and storage, all workflows completed consistently, demonstrating that backpacks enable portable, reproducible, and scalable execution of notebook workflows across diverse HPC environments.

For all the details, please check out our paper here:
Md Saiful Islam, Talha Azaz, Raza Ahmad, A D M Shahadat Hossain, Furqan Baig, Shaowen Wang, Kevin Lannon, Tanu Malik, and Douglas Thain, Backpacks for Notebooks: Enabling Containerized Notebook Workflows in Distributed Environments, IEEE Conference on eScience, pages 9, September 2025.
Project website: https://floability.github.io
Example backpacks: https://github.com/floability/floability-examples Tue, 23 Sep 2025 16:30:00 +0000Workshop on Harmonizing Python Workflows at IEEE e-Science 2025
We helped host the Workshop on Harmonizing Python Workflows at IEEE International Conference on e-Science on Monday, 15 Sep 2025.

This workshop is one component of our NSF POSE project to explore the creation of an open source ecosystem encompassing a variety of workflow technologies, including the Parsl Project (Kyle Chard at the University of Chicago), the RADICAL tools (Shantenu Jha, Rutgers University), and TaskVine (Douglas Thain, University of Notre Dame).
Workshop attendees conducted several working groups where they identified key barriers to workflow creation, deployment, and adoption; proposed activities for an open source ecosystem to support these needs; and provided feedback on the possible structure sof an ecosystem.
The organizers will be following up soon with a workshop report and draft proposal for next steps.
Thank you to everyone who participated!

Tue, 16 Sep 2025 17:10:00 +0000
Welcome Back, Colin!
This past summer, 4th year PhD student Colin Thomas completed an internship at the National Energy Research Scientific Computing Center (NERSC) located at the Lawrence Berkeley National Laboratory.
Colin worked with a team of researchers and fellow interns to develop and deploy an Inference-as-a-Service (IaaS) platform for particle physics experiments including DUNE and ATLAS. Colin organized a network of services deployed on Kubernetes and the Perlmutter supercomputer which enabled remote scientists to run their applications and perform analysis of their data. The IaaS deployment consisted of metrics collection capabilities used to profile the system, identify bottlenecks, and inform the system about the need to scale the compute resources to meet the demands of users in real-time. The team profiled multiple inference-serving technologies such as Ray Serve and NVIDIA Triton. The team effort resulted in a number of successes including tests with scientists from multiple institutions, the gathering of valuable data from the profiles of Ray and Triton, and the system prototype which can serve as an example for future work in scientific inference serving.
Colin shared with us his wonderful experience during the summer and also gave an engaging talk about the work he has done during our first team meeting. Below is the poster he created and presented. We are excited to learn new ideas and look forward to more inspiring discussions ahead.

Tue, 09 Sep 2025 20:34:00 +0000
New Semester, New Faces

The new semester is here, and we’re excited to welcome three new colleagues and roll out a clear plan for the months ahead.
New faces. Lax joins as a first-year Ph.D. student. Ryan just completed his M.S. in our lab and begins his Ph.D. this semester. Abby joins as a first-year M.S. student. We’re glad to have them on board!
Each semester we adjust our routines and schedules to keep the lab running smoothly and to support a diverse team. This semester we’re trying a few new approaches. Colin will organize our weekly team talks with a schedule planned about a month ahead, host practice sessions for conference presentations, and invite remote guests for ~30-minute Zoom talks and discussion. Jin will handle outreach and support: a short blog every Tuesday, cross-posting to LinkedIn and other channels, encouraging GitHub Discussions as a support path, and serving as the first responder for incoming technical questions. Saiful will lead the website migration to a Jekyll site on GitHub, move over most existing content, convert prior posts into native Jekyll, and switch our papers database from PHP + database to a JSON dataset. Ben, the most experienced and supportive engineer who knows every line of the codebase, will guide release engineering: review PRs and give feedback, prioritize and maintain issues, keep integration tests in working order, and plan and ship regular software releases.
Project highlights.
-
CMS Computing (Barry, Jin, Ben, Alan): keep the current TaskVine release stable and usable; share successes with the HEP community; publish the new cms-taskvine-example repository; advance an IPDPS/TPDS paper on dynamic data reduction.
-
NSF CSSI Floability (Saiful, Abby, Ben): keep the release stable, usable, and documented; attach straightforward data handling for existing applications; grow users and collaborations; build interactive visualization of notebook workflows to support troubleshooting and performance; then extend the approach to new notebook and workflow models.
-
NSF Consistency Contracts (Colin + Jin): complete and release the basic toolchain to measure, summarize, and enforce; choose baseline techniques that exploit application behavior.
-
DOE XGFabric (Ryan, Thanh): capture the full software stack; port to multiple ACCESS sites and troubleshoot issues as they appear; evaluate scale, responsiveness, and resource use; develop solutions for HPC batch queue delays.
-
NSF HARMONY: run a fall workshop on workflow collaboration with the eScience conference; capture examples across Parsl-TaskVine (astro), DaskVine (HEP), and RADICAL (xgfabric).
-
NASA-SADE (Lucas, Lax): demonstrate the simulation infrastructure at the September Year 2 review; evaluate OS scalability and performance; begin integrating native SADE components.
We’ll post a short update every Tuesday and syndicate it to LinkedIn and other channels. Questions and ideas are welcome on GitHub Discussions.
Author: Jin Zhou (jzhou24@nd.edu)
Lab PI: Douglas Thain (dthain@nd.edu)
Lab Website: https://ccl.cse.nd.edu/
GitHub: https://github.com/cooperative-computing-lab/cctools
CCL Team at GCASR 2025

Members of the CCL team traveled to Chicago, Illinois on May 8 to attend GCASR 2025 (12th Greater Chicago Area Systems Research Workshop).
CCL team members presented their work in both poster sessions.
Barry Sly-Delgado presented: Task Graph Restructuring via Function Based Annotation For Large-Scale Scientific Applications.
Colin Thomas presented: Enabling Tailored Optimizations for Scientific Workflows through I/O Tracing.
Md. Saiful Islam presented: Floability: Enabling Portable Scientific Workflows Across HPC Facilities.
Jin Zhou presented: Effectively Exploiting Node-Local Storage For Data-Intensive Scientific Workflows.
Reshaping High Energy Physics Applications Using TaskVine @ SC24
Barry Sly-Delgado presented our paper titled: "Reshaping High Energy Physics Applications for Near-Interactive Execution Using TaskVine" at the 2024 Supercomputing Conference in Atlanta, Georgia. This paper investigates the necessary steps to convert long-running high-throughput high energy physics applications to high concurrency. This included incorporating new functionality within TaskVine. The paper presents the speedup gained as changes were incorporated to the workflow execution stack for application DV3. We eventually achieve a speedup of 13X.
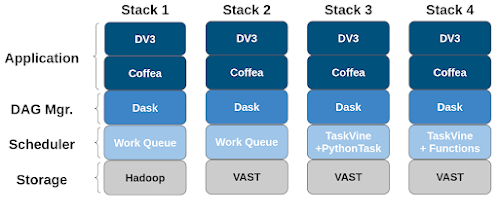
Configurations for each workflow execution stack as improvements were made.
Starting with stack 1. The first change incorporated was to the storage system where initial data sets are stored. This change showed little improvement, taking 3545s runtime to 3378s. This change is minimal as much of the data handling during application execution is related to intermediate results. Initially, with stack 1 and 2, intermediate data movement is handled via a centralized manager.
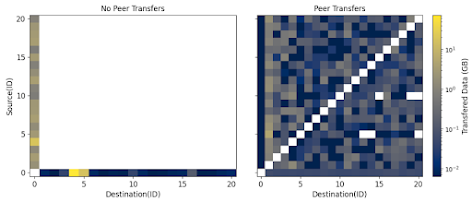
data movement during application execution between Work Queue and TaskVine. With TaskVine, the most data exchanged between any two nodes tops off around 4GB (the manger is node 0). With Work Queue the most data transferred is 40GB
Incorporated in stack 3 is a change of scheduler, TaskVine. Here, TaskVine allows for intermediate results to be stored on node-local storage and transferred between peer compute nodes. This relieves strain on the centralized manager and allows it to schedule tasks more effectively. This change drops the runtime to 730s.
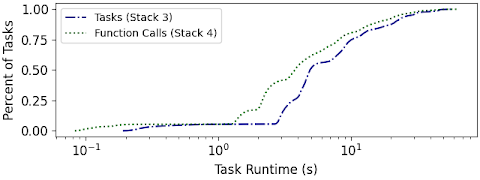
CDF of task runtimes within the application per execution paradigm. With Function Calls, individual tasks execute faster.
Our final improvement changes the previous task execution paradigm within TaskVine. Initially "PythonTasks" serialized functions along with arguments and distributed them to compute nodes to execute individual tasks. Under this paradigm, the python interpreter would be invoked for each individual task. Our new task execution paradigm, "Function Calls" stands up a persistent Python process, "Library Task", that contains function definitions that can be invoked via individual function calls. Thus, invocations of the Python interpreter are reduced from per-task to per-compute-node. This change reduces runtime to 272s for a 13X speedup from our initial configuration.
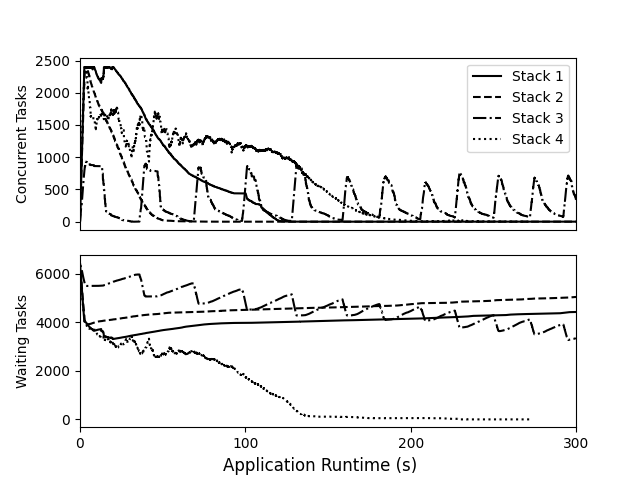
Application execution comparison between stack configurations.
Shepherd Paper at WORKS/SC 2024
Grad student Saiful Islam presented our paper on Shepherd at the 19th Workshop on Workflows in Support of Large-Scale Science at Supercomputing 2024 in Atlanta, Georgia.Shepherd is a local workflow manager that enables a fleet of actions and services to functionally behave as a single task. This allows us to seamlessly deploy these local workflows into HPC clusters at large scale. For example, consider a workflow for large-scale distributed drone simulation. This workflow includes steps such as preprocessing, creating a configuration, running simulations, and post-processing. We aim to deploy the "run simulation" step in HPC clusters at large scale. However, each "run simulation" step includes a local workflow that needs to perform specific actions and start services like Gazebo, PX4, and Pose Sender. These services and actions depend on each other’s internal states. For example, when Gazebo reaches the "ready" state, the nth PX4 service is started. Without Shepherd, the workflow becomes as shown in the following figure:

With Shepherd, the entire "run simulation" step becomes a single task of running Shepherd with a specified configuration. Shepherd starts the required actions and services, manages dependencies, and ensures the graceful shutdown of services when they are no longer needed. This enables complex workflows of actions and services to be seamlessly integrated with a larger distributed workflow manager, as shown in the following figure:

The paper discusses the architecture and design principles of Shepherd, showcasing its application to large-scale drone simulations and integration testing.
Shepherd uses a YAML-based workflow description to define tasks, dependencies, and execution conditions. It monitors logs and file generation to infer internal states and manage lifecycles effectively. Additionally, it generates three visualizations post-execution for debugging and documentation. For instance, the figure below illustrates a timeline of a 100-drone simulation distributed across 25 nodes, with each Shepherd instance managing 4 drones. A zoomed-in view highlights how components execute at varying times across nodes, which becomes challenging without awareness of service readiness. Shepherd simplifies this with YAML-based configurations and internal state tracking.

For all the details, please check out our paper here:
- Md Saiful Islam, Douglas Thain, Shepherd: Seamless Integration of Service Workflows into Task-Based Workflows through Log Monitoring, 19th Workshop on Workflows in Support of Large-Scale Science at ACM Supercomputing, pages 1-8, November, 2024.
Tue, 07 Jan 2025 23:50:00 +0000
Data Pruning Mechanism in Daskvine
In our recent work, we introduced a file pruning technique into DaskVine to address challenges in managing intermediate files in DAG-based task graphs. This technique systematically identifies and removes intermediate stale files—those no longer needed by downstream tasks—directly from worker nodes. By freeing up storage in real time, file pruning not only enables workers to process more computational tasks with limited disk space but also makes applications feasible that were previously constrained by storage limitations.
Specifically, the pruning algorithm monitors task execution in the graph, categorizing tasks as waiting or completed. Once a task finishes, it prunes its parent tasks’ output files if all dependent child tasks are done and immediately submits dependent tasks for execution when their inputs are ready.
To evaluate the effectiveness of our file pruning technique, we utilized four key metrics:
File Retention Rate (FRR): FRR represents the ratio of a file's retention time on storage to the total workflow execution time. It indicates how long files remain in storage relative to the workflow's progress. A shorter FRR reflects more efficient pruning. The following figures show the FRR over all intermediate files, each producing by one specific task. The left graph (FCFS) shows higher and more uniform FRR, where files remain on storage for a significant portion of the workflow. In contrast, the right graph (FCFS with Pruning) shows a clear reduction in FRR for most files, which proves that pruning reduces storage usage by removing stale files promptly.

Accumulated Storage Consumption (ASC) and Accumulated File Count (AFC): ASC measures the total amount of storage consumed across all worker nodes throughout the workflow execution, while AFC tracks the total number of files retained across all worker nodes during the workflow. In the following, the left graph shows that both ASC and AFC steadily increase throughout the workflow, peaking at 1082.94 GB and 299,787 files, respectively. This highlights significant storage pressure without pruning. The right graph demonstrates dramatic reductions, with peaks at 326.78 GB for ASC and 80,239 files for AFC. This confirms that pruning effectively reduces storage consumption and file count by promptly removing unnecessary intermediate files.

Worker Storage Consumption (WSC): WSC reflects the storage consumption of individual workers at any point during the workflow execution. It helps assess the balance of storage usage among workers. The left graph shows a peak WSC of 24.95 GB, indicating high storage pressure on individual workers. In contrast, the right graph shows a significantly lower peak of 7.07 GB, demonstrating that pruning effectively reduces storage usage per worker and balances the load across the cluster.

Mon, 09 Dec 2024 16:10:00 +0000
TaskVine + Parsl Integration
The Cooperative Computing Lab team has an ongoing collaboration with the Parsl Project, maintaining the TaskVine Executor for use with the Parsl workflow system. Using the TaskVine executor involves expressing an application using the Parsl API, where tasks are created and managed using the Parsl Data Flow Kernel. Tasks are passed to the executor which makes final scheduling decisions.
The TaskVine executor offers a number of features involving data management and locality scheduling. The distinction between the TaskVine executor and other available executors is largely the awareness of data dependencies and the use of node-local storage. There are a number of features to the TaskVine executor that have been recently added or are in progress.
One prominent new feature is the extension of TaskVine function invocations to the TaskVine executor. Function invocations reduce the overhead of starting new tasks, considerably benefiting workflows with many short-running tasks. TaskVine function calls have been described with greater detail in this article by Thanh Son Phung. This benefit to task startup latency is especially useful for the Parsl/TaskVine executor, since Parsl is a python-native application. Normally each task must consider starting a new python process and loading all modules used by Parsl as well as the user's application. With short running tasks, such as ones resembling quick function calls, the overhead of loading this information from shared storage, or transferring it to local storage and starting a new python process can be greater than the running time of the task itself. TaskVine function invocations allow this runtime environment information to be effectively cached, and multiple tasks to run sequentially in the same python process. A molecular dynamics application using the Parsl/TaskVine executor is shown below, with L1 being simple tasks, and L2 using TaskVine function invocations.

Other new changes to the TaskVine executor include the addition of the tune_parameters to the executor configuration. TaskVine is highly configurable by users to suit the needs of their particular application or cluster resources. Adding this option to the TaskVine executor allows users to automatically replicate intermediate data, configure the degree of replication desired, disable peer transfers, and several in-depth scheduling options which fit different specific task dispatch and retrieval patterns.
Accelerating Function-Centric Applications via Reusable Function Context in Workflow Systems
Modern applications are increasingly being written in high-level programming languages (e.g., Python) via popular parallel frameworks (e.g., Parsl, TaskVine, Ray) as they help users quickly translate an experiment or idea into working code that is easily executable and parallelizable on HPC clusters or supercomputers. Figure 1 shows the typical software stack of these frameworks, where users wrap computations into functions, which are sent to and managed by a parallel library as a DAG of tasks, and these tasks eventually are scheduled by an execution engine to execute on a remote worker node.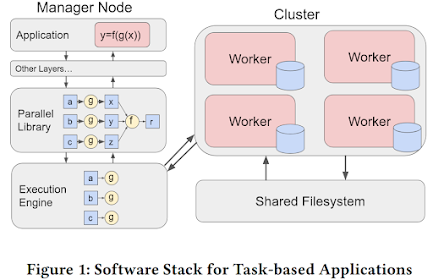
A traditional way to execute functions remotely is to translate them into executable tasks by serializing functions and their associated arguments into input files, such that these functions and arguments are later reconstructed on remote nodes for execution. While this way fits function-centric applications naturally into well understood task-based workflow systems, it brings a hefty penalty to short-running functions. A function now takes extra time for its states to be sent and reconstructed on a remote node which are then unnecessarily destroyed at the end of that function’s execution.
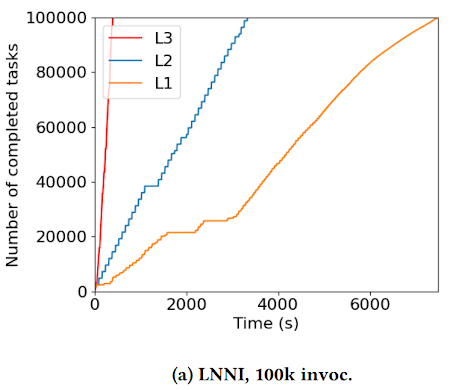
At HPDC 2024, graduate students Thanh Son Phung and Colin Thomas proposed the idea that function contexts, or states, should be decoupled from function’s actual execution code. This removes the overhead of repeatedly sending and reconstructing a function’s state for execution, and allows functions of the same type to share the same context. The rest of the work then addresses how a workflow system can treat a function as a first-class citizen by discovering, distributing, and retaining such context from a function. Figure below shows the execution time of the Large-Scale Neural Network Inference application, totaling 1.6 million inferences separated into 100k tasks, with increasing levels of context sharing (L1 is no sharing, L3 is maximum sharing). Decoupling the inference function’s context from the inference massively reduces the execution time of the entire workflow by 94.5%, from around 2 hours to approximately 7 minutes.
An interested reader can find more details about this work in the paper:Thanh Son Phung, Colin Thomas, Logan Ward, Kyle Chard, and Douglas Thain. 2024. Accelerating Function-Centric Applications by Discovering, Distributing, and Retaining Reusable Context in Workflow Systems. In Proceedings of the 33rd International Symposium on High-Performance Parallel and Distributed Computing (HPDC '24). Association for Computing Machinery, New York, NY, USA, 122–134. https://doi.org/10.1145/3625549.3658663
Mon, 11 Nov 2024 16:07:00 +0000
A New Visualization Tool for TaskVine Released
We released a web-based tool to visualize runtime logs produced by TaskVine, available on Github. Using this tool involves two main steps. First, the required data in CSV format must be generated for the manager, workers, tasks, and input/output files. After saving the generated data in the directory, users can start a port on their workstation to view detailed information about the run. This approach offers two key advantages: the generated data can be reused multiple times, minimizing the overhead of regeneration, and users can also develop custom code to analyze the structured data and extract relevant insights.
For example, the first section describes the general information of this run, including the start/end time of the manager, how many tasks are submitted, how many of them succeeded or failed, etc.

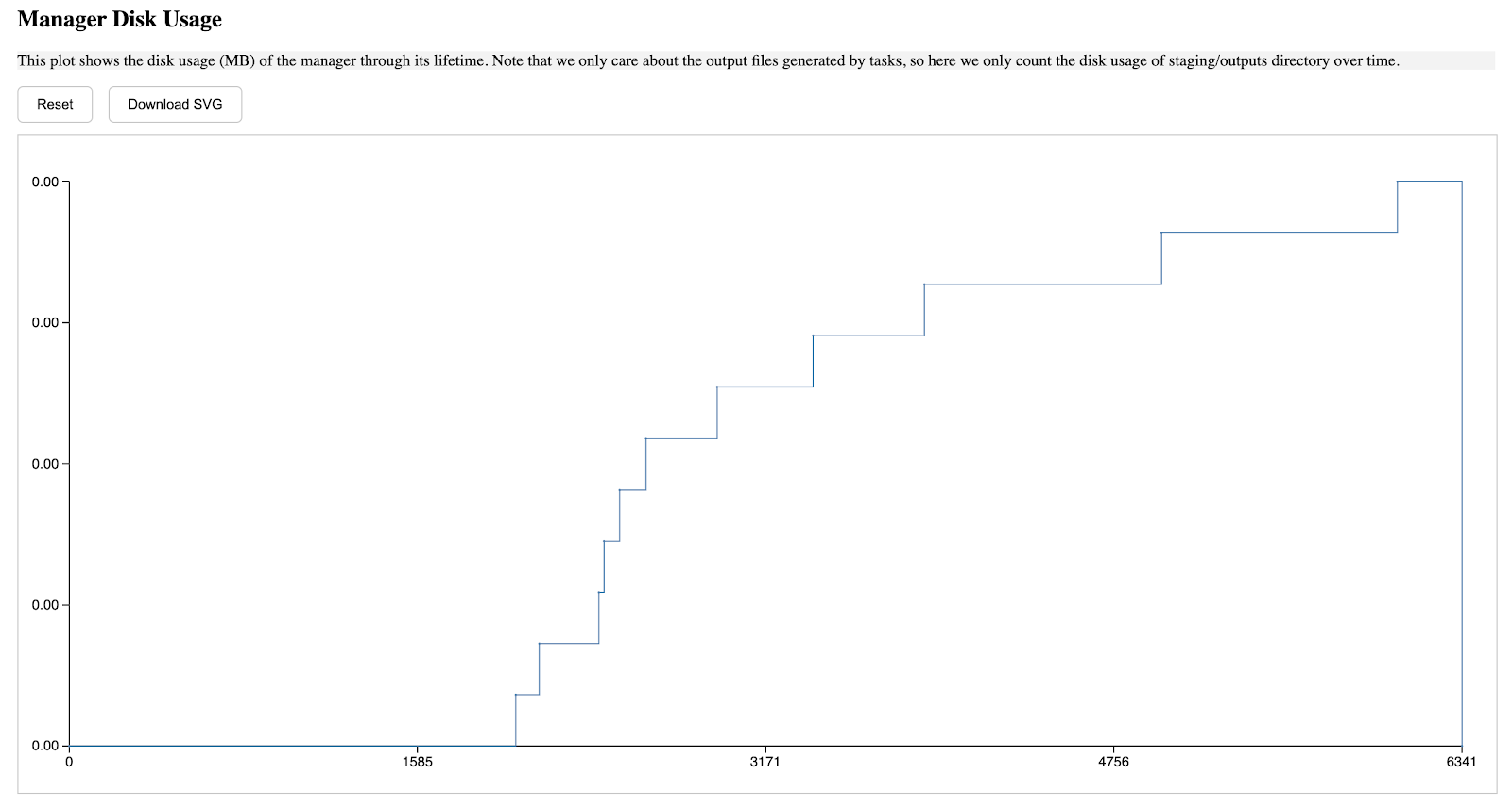
The second section describes the manager's storage usage through its lifetime, the a-axis starts from when the manager is started, and ends when the manager is terminated, the y-axis is in MB unit, and such pattern is applied to all diagrams in this report.
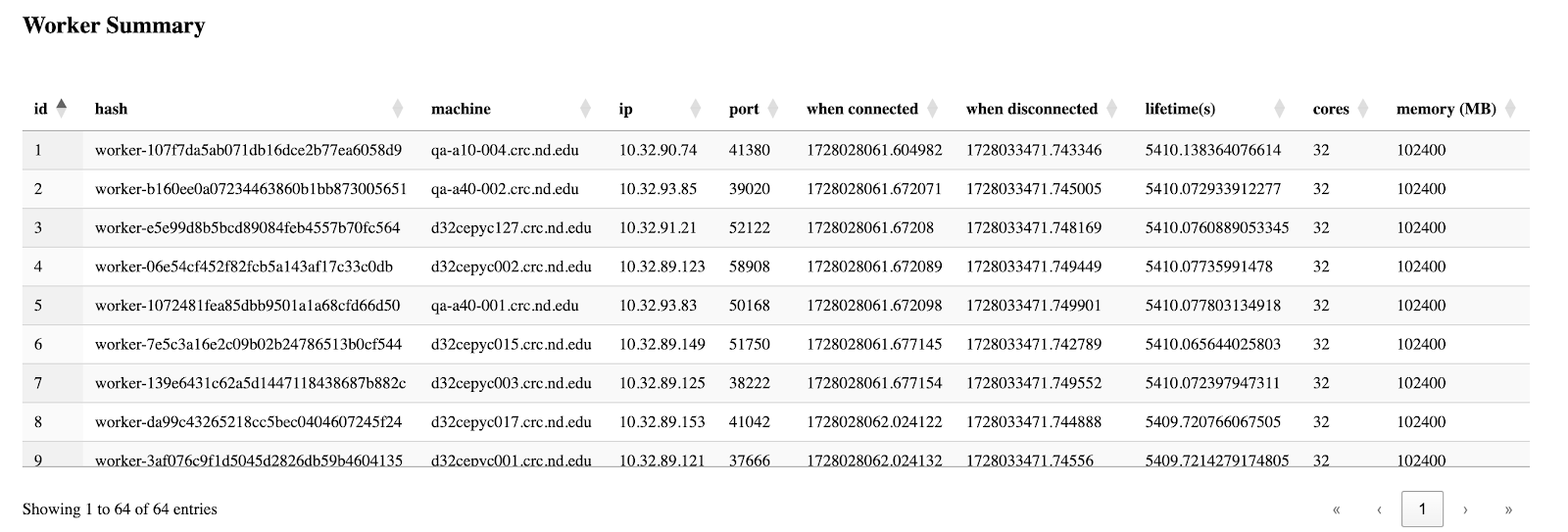
The third section is the table of all workers' information, which is basically grabbed from the csv files from the backend, but this enables users to easily sort by their interested columns.
The fourth section is the storage consumption among all workers. Several buttons in the top are provided, to turn the y-axis to a percentage unit, or to highlight one worker that is of interest.

The fifth section is the number of connected workers throughout the manager's lifetime, hovering the mouse on a point shows the information of a connected/disconnected worker.

The sixth section shows the number of concurrent workers throughout the manager's lifetime.

The seventh section is table of completed/failed tasks and their information.
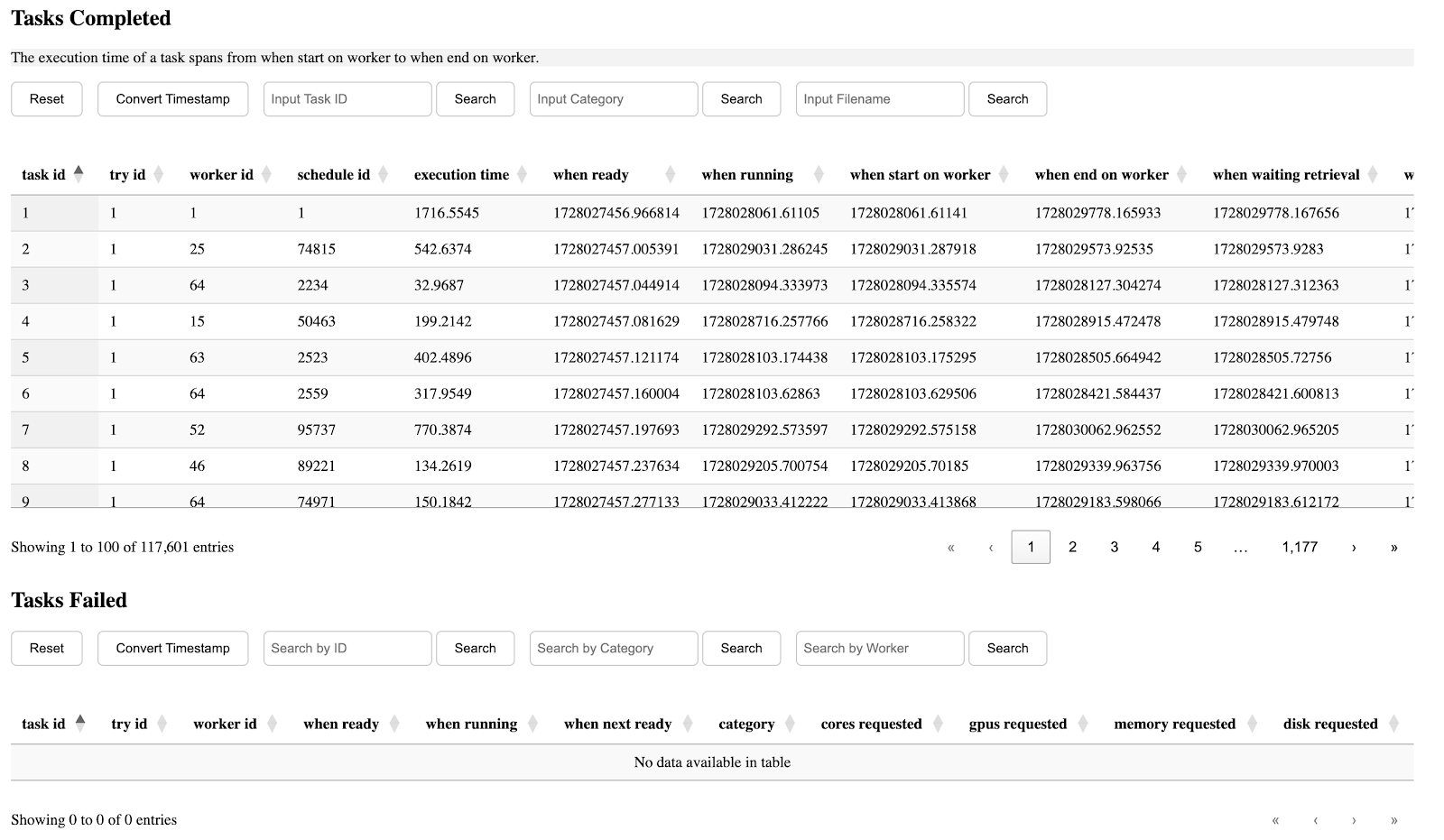
The eighth section is the execution time distribution of different tasks. Those with a lower index on the left side of the x-axis are submitted earlier, while those on the right are submitted later. A CDF can be seen by clicking the button on the top.

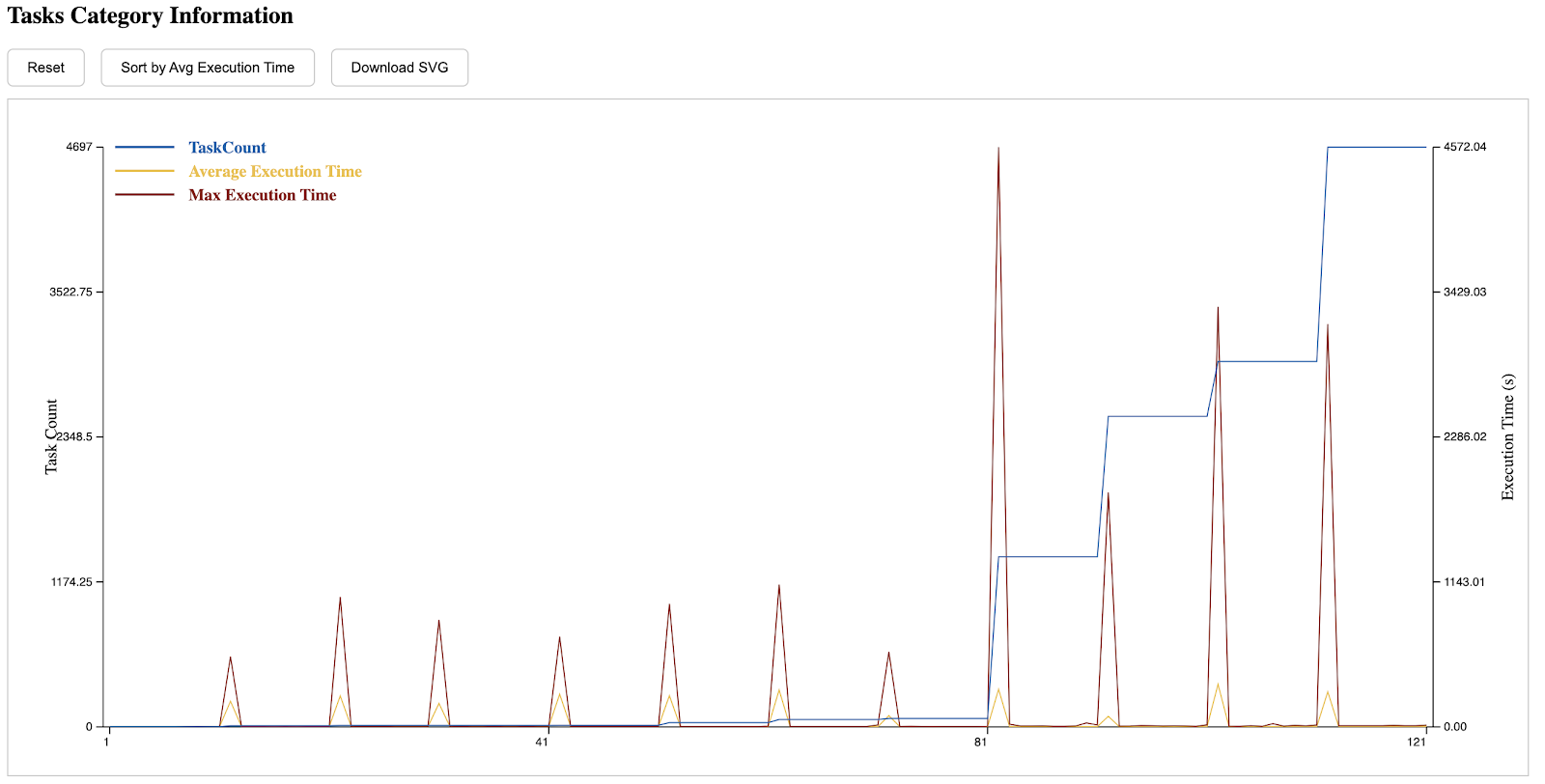
The tenth section demonstrates the general runtime distribution of tasks, the y-axis is the worker-slot pair. In the following example, we have 64 workers and each with 20 cores. One can zoom in the diagram and hover to see the detailed information of one task, which is particularly useful when examining outliers.


The last section is about the structure of the compute graphs. Nodes in the graph are tasks with an index label, while edges are the dependencies between input/output files of tasks. Weights on those task->file edges are the execution time of tasks, while those on the file->task edges are the waiting time starting from when a file is produced to when it is consumed by a consumer task.

This tool works well under the scale of hundreds of thousands of tasks, but for large runs, which may have millions of tasks, the online visualization tool may be unable to process such amount of data because the data transferring bottleneck between backend and frontend. Under such case, or just for convenience, we recommend the users to use tools under the pyplot directory, which is more lightweight and uses traditional matplotlib and seaborn to draw diagrams. Detailed explanations are provided under the README file.
Mon, 04 Nov 2024 23:34:00 +0000Integrating TaskVine with Merlin
Graduate student, Barry Sly-Delgado, completed a summer internship onsite at Lawrence Livermore National Laboratory where he worked on integrating TaskVine with Merlin, an executor for machine learning workflows. Barry worked as a member of the WEAVE team under Brian Gunnarson and Charles Doutriaux.
Previously, Merlin used Celery to distribute tasks across a compute cluster. With TaskVine's addition, utilization of in-cluster resources (bandwidth, disk) is available for workflow execution. Existing Merlin specifiacations can use TaskVine as a task scheduler with little change to the specification itself.
Merlin works with TaskVine by utilizing the Vine Stem, a DAG manager that borrows the concepts of groups and chains to create workflows from Celery. With this, the Vine Stem sends tasks (Merlin Steps) to the TaskVine manager for execution. Execution of these tasks eventually create a directory hierarchy that previous Merlin workflows already do. In addition to the workflow specification, a Merlin Specification contains specifications for starting workers, which are submitted via a batch system (HTCondor, UGE,Slurm)
Architecture of Merlin with TaskVine
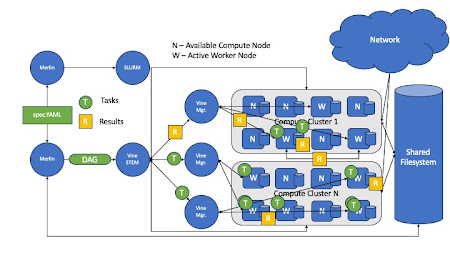

Sample Merlin Specification Block With TaskVine as Task Server
The TaskVine task server option will be included in an upcoming release of Merlin. We would be happy to find more use cases so please check it out once released!
Wed, 16 Oct 2024 19:14:00 +0000
Introducing Shepherd: Simplifying Integration of Service Workflows into Task-Based Workflows
We are pleased to announce the release of Shepherd, an open-source tool designed to streamline the integration of service workflows into task-based workflows. Shepherd enables users to manage applications that require dynamic dependencies and state monitoring, ensuring that each component in a workflow starts only after its dependencies have reached specified states.
What Is Shepherd?
Shepherd is a local workflow manager and service orchestrator that launches and monitors applications according to a user-defined configuration. It bridges the gap between persistent services and traditional task-based workflows by treating services as first-class tasks. This approach allows for seamless integration of services that run indefinitely or until explicitly stopped, alongside actions that run to completion.
By analyzing log outputs and file contents, Shepherd tracks the internal states of tasks without the need to modify their original code. This capability is particularly useful when integrating persistent services into workflows where tasks may depend on the internal states of these services rather than their completion.

Key Features
- Services as Tasks: Treats both actions (tasks that run to completion) and services (persistent tasks that require explicit termination) as tasks within a workflow, allowing integration into task-based workflow managers.
- Dynamic Dependency Management: Initiates tasks based on the internal states of other tasks, enabling complex state-based dependencies. Supports both "any" and "all" dependency modes for flexible configurations.
- State Monitoring: Continuously monitors the internal state of each task by analyzing standard output and specified files, updating states in real-time based on user-defined patterns.
- Graceful Startup and Shutdown: Ensures tasks start only when their dependencies are met and provides controlled shutdown mechanisms to maintain system stability and prevent data loss.
- Logging and Visualization: Generates comprehensive logs and state transition data, aiding in performance analysis and debugging.
- Integration with Larger Workflows: By encapsulating service workflows into single tasks, Shepherd enables integration with larger distributed workflow managers, enhancing workflow flexibility and reliability.
Documentation and Source Code
For detailed documentation, examples, and source code, please visit the Shepherd GitHub Repository.
Mon, 14 Oct 2024 13:55:00 +0000
TaskVine at ParslFest 2024
On September 26-27 members of the CCL team attended ParslFest 2024 in Chicago, Illinois to speak about TaskVine and connect with our ongoing collaborators at the Parsl Project.
Grad student Colin Thomas delivered a talk titled "Parsl/TaskVine: Interactions between DAG Manager and Workflow Executor."
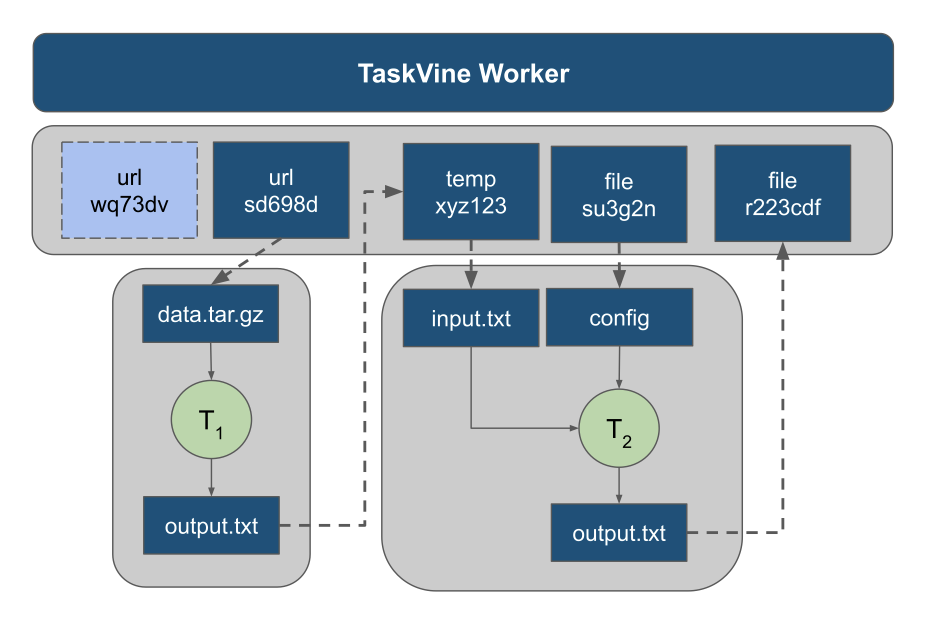
The talk highlighted recent developments in the implementation of TaskVine Temporary Files within the Parsl/TaskVine Executor. Temporary files allow users to express intermediate data in their workflows. Intermediate data are items produced by tasks, and consumed by tasks. With the use of TaskVine Temporary Files, this intermediate data will remain in the cluster for the duration of a workflow. Keeping this data in the cluster can benefit a workflow that produces intermediate files of considerable quantity or size. 
In addition to adding temporary files to the Parsl/TaskVine executor, the talk presented ongoing work on the development of intermediate data-based "task-grouping" in TaskVine and extending it to the TaskVine executor. The concept of grouping related tasks may sound familiar to Pegasus users or other pre-compiled DAG workflow systems. Currently task-grouping is focused on scheduling sequences of tasks which contain intermediate dependencies. The desired outcome is for TaskVine to interpret pieces of the DAG to identify series of sequentially-dependent tasks in order to schedule them on the same worker, such that intermediate data does not need to travel to another host. Extending this capability to Parsl brought about an interesting challenge which questioned the relationship between DAG manager and executor.
The problem is that in the typical workflow executor scheme, such as with Parsl or Dask, the executor will receive tasks from the DAG manager only when they are ready to run. In order for TaskVine to identify sequentially dependent tasks it needs to receive tasks which are future dependencies of those that are ready to run.
The solution to this came about as a special implementation of a Data Staging Provider in Parsl, which is the mechanism for determining whether data dependencies are met. When the TaskVine staging provider encounters temporary files, it will lie to the Parsl DFK (Data Flow Kernel) in saying the file exists. Therefore Parsl will send all temporary-dependent tasks to TaskVine without waiting for the files to actually materialize. This offers TaskVine relevant pieces of the DAG to inspect and identify dependent sequences.
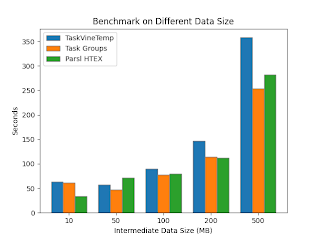

The impact of task grouping was evaluated on a benchmark application. Each benchmark run consists of 20 task sequences of variable length. Each task in a sequence produces and consumes an intermediate file of variable size. The effect on running time is shown while adjusting these parameters. Grouping tasks was shown to benefit the performance across a variety of sequence lengths, and when intermediate data size exceeds 500MB.
In addition to the positive benchmark results, this talk promoted discussion about this non-traditional interaction between DAG manager and executor. The DAG manager has insight into future tasks and their dependencies. The executor possesses knowledge about data locality. Combining this information to make better scheduling decisions proved to have some utility, yet it challenged the typical scheme of communication between Parsl and the TaskVine executor.
Predicting Resources of Tasks in Dynamic Workflows with Bucketing Algorithms at IPDPS 2024
Thanh Son Phung will present Adaptive Task-Oriented Resource Allocation for Large Dynamic Workflows on Opportunistic Resources at the International Parallel and Distributed Processing Symposium 2024.
Non-technical users running dynamic workflows usually have no knowledge of resource (e.g., cores, memory, disk, GPUs) consumption of tasks within a given workflow. However, this omission of information can bring substantial performance degradation as it is important for workflow execution engines to know the exact amount of resources each task takes and make scheduling decisions accordingly.

In the figure above, an application submits many invocations of two functions: f and g. The invocations are transformed into tasks through many layers and scheduled on worker nodes. Note that the resource specification of each task may be unknown.
Since a manual resource specification for each task is unreliable, an automated solution is required. Such solution needs to be (1) general-purpose (can work with any workflow), (2) prior-free (no prior information is used as workflows may change over each run), (3) online (collect information and run as the workflow runs), and (4) robust (performant on many distributions and unexpected changes). The below figure shows two workflows: ColmenaXTB (top row) and TopEFT (bottom row). Resource types range from core, memory, disk, and execution time from left to right.

To predict resources of each task, we first collect resource records of completed tasks, group them into buckets of tasks based on their similarity in resource consumption, and probabilistically choose a bucket to allocate the next task, as shown in the example below.

We introduce two bucketing algorithms, namely Greedy Bucketing and Exhaustive Bucketing, which share the same principle of resource prediction and only differ in the way buckets of tasks are computed.

We compare the two algorithms with five alternative algorithms on 7 workflows with 3 resource types. Results show that Greedy Bucketing and Exhaustive Bucketing consistently outperform other algorithms by yielding the highest resource efficiency in the majority of cases.
Further details can be found in the paper:
- Thanh Son Phung, Douglas Thain, Adaptive Task-Oriented Resource Allocation for Large Dynamic Workflows on Opportunistic Resources, IEEE International Parallel and Distributed Processing Symposium (IPDPS), pages 12, May, 2024.
TaskVine at the HEP Analysis Grand Challenge
Barry Sly-Delgado and Ben Tovar recently presented our work on transforming high energy physics data analysis applications into near-interactive execution at the IRIS-HEP Analysis Grand Challenge Demo Day.
By using the TaskVine framework, we have enabled the transformation of a production physics data analysis application (DV3), reducing execution time from over an hour to just over three minutes
on about four thousand cores of a campus HPC cluster.

Advancements to hardware and software components in an application stack aid in the facilitation of application reshaping. With application reshaping, we are transitioning applications from long running to near-interactive. While improvements to hardware and low-level software improvements can produce measurable speedup, significant speedup is obtainable via optimization to the scheduling and execution layers of the distributed application.

TaskVine, our latest workflow technology, makes use of in-cluster bandwidth and disk to mitigate data movement by enabling peer transfers between worker nodes within a compute cluster. TaskVine is currently used to run a variety of custom data analysis workflows written by the CMS physics group at Notre Dame. These applications are written in high-level Python, making use of standard technologies like Numpy, Coffea, and Dask in order to generate large task graphs. TaskVine then takes that graph and deploys it into the cluster.

Our previous versions of these physics applications utilized our earlier WorkQueue executor. TaskVine improves upon the earlier system in two distinct ways:
First - While Work Queue only transfers data between the manager and workers, TaskVine transfers data directly between peer workers. Peer transfers relieve pressure on the manager to distribute data dependencies to worker nodes within the cluster. For workflows that generate large amounts of intermediate data, this can be extremely costly to scheduling performance, because the manager sends and receives large amounts of data.
This animation shows the data transfer throughout an application when using Work Queue:

While this animation shows the data transfers between all nodes of the cluster when using TaskVine:

Second - TaskVine extends the notion of task execution into a serverless model. Python code in the analysis application can be defined once as a LibraryTask which is deployed into the cluster. Then, lightweight FunctionCall tasks can invoke the code in the LibraryTask without constantly restarting the Python environment. In this modem the python interpreter is now invoked once per worker.
This graph compares the distribution of execution times between normal tasks and serverless function calls, improving the lower limit of execution time by an order of magnitude.

These four graphs show the overall performance of the data analysis application through four improvement states: first with Work Queue and Hadoop, then with Work Queue and VAST (new HPC filesystem), then TaskVine with peer transfers, and finally TaskVine with serverless execution. Overall, this transforms a one hour workflow into a 3 minute workflow.


CCTools 7.8.0 released
We are pleased to announce the release of version 7.8.0 of the
Cooperative Computing Tools from the University of Notre Dame, including
TaskVine, Work Queue, Makeflow, Parrot, Chirp, and other software.
Docs: https://cctools.readthedocs.io/
Download: http://ccl.cse.nd.edu/software/download
Forum: http://ccl.cse.nd.edu/community/forum
This is a feature release that primarily improves the scalability and performance of TaskVine:
Main Features:
TaskVine dask serverless execution (i.e., function calls instead of tasks) to reduce overheads, by @BarrySlyDelgado
TaskVine improvements to library and remove function calls dispatch, by @tphung3
TaskVine temporary file replication, by @colinthomas-z80
TaskVine automatic deletion of ancillary files (unlink_when_done) during workflow execution, by @btovar
TaskVine use of memory buffers to serialize functions, by @btovar
TaskVine cleanup of factory, worker information and stats, by @dthain
Fix TaskVine memory leak in python bindings, by @JinZhou5042
Improved Parsl integration with TaskVine and documentation, by @colinthomas-z80
Full Changelog
Distant Futures at SC 2023
Grad student Barry Sly-Delgado presented his recent work on "Minimizing Data Movement Using Distant Futures" at the research poster session at Supercomputing 2023:


Futures are a widely used concept for organizing concurrent computations. A Future is a variable that represents the result of a function call whose computation may still be pending. When evaluated, the future blocks (if needed) until the result becomes available. This allows for the easy construction of highly parallel programs.
Barry developed a future based execution model for the TaskVine workflow execution system. This allows one to submit and chain function calls written in plain Python. TaskVine then schedules the tasks throughout the cluster, providing a variety of data management services.

However, at very large scales, a natural bottleneck is the return of computed values back to the original manager. To overcome this, we introduce the notion of Distant Futures in which a value is not only pending in time, but potentially left in the cluster on a remote node. Tasks requiring this value can then be scheduled to the same node, or to transfer the value within the cluster, rather than bringing it back home.
Combining distant futures with asynchronous transfer provides significant benefits for applications that are bottlenecked in data transfer, as our results show:

For more details, check out the poster and short paper here:
- Barry Sly-Delgado and Douglas Thain, Poster: Minimizing Data Movement Using Distant Futures ACM/IEEE Supercomputing, November, 2023.
Maximizing Data Utility at HPPSS/SC 2023
Thanh Son Phung presented Maximizing Data Utility for HPC Python Workflow Execution at the High Performance Python for Science at Scale workshop at Supercomputing 2023.
This paper describes how the Parsl workflow system integrates with the TaskVine workflow executor in order to run complex Python environments at scale. Parsl provides a function-oriented programming model, generates a large number of tasks to be executed in a graph, and then passes the tasks down to TaskVine for execution on a cluster:

A significant challenging in scaling up Python applications is managing the large body of dependencies in the form of Python modules, system libraries, and so forth, that must be loaded every time a process starts. At very large scales, the cost of pulling all of these resources from a shared parallel filesystem can become a significant bottleneck. (Typically, parallel filesystems are optimized for the delivery of a few large files, rather than a large number of small files.)
TaskVine addresses the problem by exploiting the storage capability of each execution node, which is typically equipped with a local SSD for temporary storage. The TaskVine workers pull data into local storage, and then share files with each other (at the direction of the manager), resulting in less pressure on the shared filesystem, and overall speedups that improve with scale.
This technique was evaluated on a machine learning application requiring a (quite common) 4.4GB of software scaling up from 32 nodes x 16 cores to 256 nodes x 16 cores on a campus HPC cluster. At small scales, TaskVine provides a modest improvement, but as scale increases, the separation becomes more distinct:

The distribution of task execution times shows that the TaskVine variants (bottom row) shows that tasks run more consistently with fast individual execution times, once data is distributed across the cluster.

For the full details, see our paper here:
|
TaskVine Paper at WORKS/SC 2023
Barry Sly-Delgado presented our overview paper on TaskVine at the Workshop on Workflows in Support of Large Scale Science at Supercomputing 2023 in Denver, Colorado.
TaskVine is a system for executing data intensive workflows on large clusters. These workflows ma consist of thousands to millions of individual tasks that are connected together in a graph structure like this:

When executing this sort of workflow in a cluster, the movement of data between tasks is often the primary bottleneck, especially if each item must flow back and forth between a shared filesystem.
The key idea of TaskVine is to exploit the local storage available on each node of a cluster to relieve much of the load placed on a shared filesystem. A running TaskVine system consists of a manager process that coordinates data transfers between workers, like this:

A workflow in TaskVine is expressed in in Python by declaring the data assets needed by the workflow as file objects, and then connecting them to the namespace of each task to execute:

When the tasks execute on each worker, the various data objects are cached on local storage, and then linked into the execution sandbox of each running task. This provides the opportunity for data to be shared between tasks without moving data to/from the shared filesystem:
This paper gives three examples of applications constructed using TaskVine. TopEFT is a physics data analysis application built using Coffea+TaskVine, Colmena-XTB is a molecular modeling application built using Parsl+TaskVine, and BGD is a model training application built using TaskVine alone. Each of these codes is able to scale to 400-1000 nodes running on 1000-27000 cores total.

For all the details, please check out our paper here:
- Barry Sly-Delgado, Thanh Son Phung, Colin Thomas, David Simonetti, Andrew Hennessee, Ben Tovar, Douglas Thain, TaskVine: Managing In-Cluster Storage for High-Throughput Data Intensive Workflows, 18th Workshop on Workflows in Support of Large-Scale Science, November, 2023. DOI: 10.1145/3624062.3624277
CCTools 7.7.0 Released
We are pleased to announce the release of version 7.7. of the Cooperative Computing Tools from the University of Notre Dame, including TaskVine, Work Queue, Makeflow, Parrot, Chirp, and other software.
- Docs: https://cctools.readthedocs.io/
- Download: http://ccl.cse.nd.edu/software/download
- Forum: http://ccl.cse.nd.edu/community/forum
This is a feature release that primarily improves the scalability and performance of TaskVine:
Main Features:
- TaskVine workers now perform transfers asynchronously while communicating with manager, by @BarrySlyDelgado
- TaskVine Serverless function execution simplified and accelerated by @tphung3
- Improved integration of Parsl and TaskVine by @tphung3
- Improved scheduler performance in Work Queue and TaskVine by @colinthomas-z80
- Multiple improvements to reliability at scale with physics applications by @btovar
- Change to API for cancelling tasks: all tasks are now returned by
vine_waitby @dthain - Change to serverless resource management: functions consume library resources by @dthain
- Improved visualization by @JinZhou5042
- New environment.yml for development dependencies by @thieber22
Full Changelog: nightly...release/7.7.0
Tue, 03 Oct 2023 17:15:00 +0000CCTools 7.6.1 Released
We are pleased to announce the release of version 7.6.1
of the Cooperative Computing Tools from the University of Notre Dame,
including TaskVine, Work Queue, Makeflow, Parrot, Chirp, and other software.
Docs: https://cctools.readthedocs.io/
Mon, 24 Jul 2023 20:06:00 +0000
Download: http://ccl.cse.nd.edu/software/download
Forum: http://ccl.cse.nd.edu/community/forum
This release introduces several bug fixes and minor improvements to the TaskVine workflow executor and Work Queue scheduler.
TaskVine:
Forum: http://ccl.cse.nd.edu/community/forum
This release introduces several bug fixes and minor improvements to the TaskVine workflow executor and Work Queue scheduler.
TaskVine:
- Fix bug in task scheduling that would result in manager hanging. (Colin Thomas)
- Fix bug in execution of Dask task graphs that would lead to a hang. (Ben Tovar)
- Fix bug in deployment of serverless LibraryTask. (Thanh Phung)
- Add option to specify Python package dependencies inline. (Barry Sly-Delgado)
- Add visualization of task graph and documentation. (Douglas Thain)
Work Queue:
- Fix bug in execution of Dask task graphs that would lead to a hang. (Ben Tovar)
- Fix bug in deployment of serverless LibraryTask. (Thanh Phung)
- Add option to specify Python package dependencies inline. (Barry Sly-Delgado)
- Add visualization of task graph and documentation. (Douglas Thain)
Work Queue:
- Fix bug in task scheduling that would result in manager hanging. (Colin Thomas)